ETIOLOGÍA DE LA OSTEOGÉNESIS IMPERFECTA
La OI es causada por defectos en una proteína llamada colágeno tipo 1 o defectos relacionados con ella. El colágeno es un bloque de construcción esencial del cuerpo. El cuerpo usa el colágeno tipo 1 para hacer huesos más fuertes y para construir tendones, ligamentos, dientes y el blanco de los ojos.
Algunos cambios genéticos, o mutaciones, causan los defectos en el colágeno. Hay mutaciones en diversos genes que pueden provocar la OI. Alrededor del 80%–90% de los casos de OI se debe a mutaciones autosómicas dominantes en los genes del colágeno tipo 1, COL1A1 and COL1A2. Las mutaciones en uno u otro de estos genes hacen que el cuerpo produzca colágeno con una formación anormal o en muy poca cantidad. Las mutaciones en estos genes causan la OI tipos I a IV.
El resto de los casos de OI (tipos VI-XI) son el resultado de mutaciones autosómicas recesivas en cualquier de los seis genes (SERPINF1, CRTAP, LEPRE1, PPIB, SERPINH1, y FKBP10) que codifican las proteínas que ayudan a producir colágeno. Estas mutaciones también hacen que el cuerpo produzca demasiado poco colágeno o colágeno con una formación anormal..jpg)
Estos cambios genéticos son hereditarios, o transmitidos de los progenitores a sus hijos; las personas que tienen OI nacen con la enfermedad. Sin embargo, en algunos casos, la mutación genética no se hereda sino que ocurre luego de la concepción.
DESARROLLO DE LA ENFERMEDAD
La OI es una enfermedad rara. Su incidencia se estima entre 1:10.000 y 1:15.000. Esta estimación es un límite inferior, ya que las formas livianas de la enfermedad frecuentemente no se diagnostican. La OI ocurre en todas las razas y es independiente de género.
Solamente un 0.008% de la población mundial está afectada por la OI. Esto significa que en la actualidad hay unos 0.5 Millones de personas con OI en el mundo.
En España podría haber un mínimo de 2700 afectados por alguno de los tipos de OI.
SINTOMAS
El diagnóstico de una persona con OI suele ser fundamentalmente por exploración y basarse en alguna o varias de las siguientes manifestaciones:
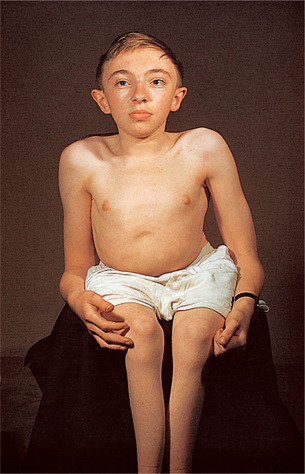
- Fragilidad ósea (los huesos se fracturan incluso sin causa aparente).
- Cara en forma triangular (puesto que el cráneo crece empujado por el encéfalo, mientras que la mandíbula no tiene qué la haga crecer).
- Escleróticas (lo blanco de los ojos) azules o grises.
- Sordera progresiva, habitualmente en la edad adulta.
- Dentinogénesis imperfecta (decoloración y fragilidad en los dientes).
- Tono de voz agudo.
- Estatura baja.
- Tendencia a magullarse la piel y aparición frecuente de "cardenales".
- Músculos débiles.
- Articulaciones laxas.
- Escoliosis.
- Deformidades óseas (extremidades superiores, inferiores, pecho y cráneo).
- Estreñimiento.
- Sudoración excesiva.
- Coeficiente intelectual medio- alto.
- Tono vital con tendencia al optimismo y la euforia.
Lo habitual es que no se den todas estas manifestaciones al mismo tiempo en un afectado de OI.
DIAGNOSTICO
El diagnóstico de O.I. se puede establecer mediante ecografía perinatal, que permite detectar afectos alrededor de la 16ª semana de gestación.
Puede llegarse al diagnóstico mediante la demostración de la síntesis de cadenas pro-alfa anormales o de la secuenciación del DMA en las vellosidades coriónicas obtenidas por biopsia entre la 8 y 12 semanas de gestación.
Ninguno de estos diagnósticos es fácil de realizar, y alguno puede tener ciertos riesgos que deben ser evaluados por los especialistas médicos.
TRATAMIENTO
La enfermedad dura toda la vida del paciente, requiriéndose cuidados y control desde antes del nacimiento (si es posible y si está diagnosticado): estudio genético y de fertilización, cuidados obstétricos, y controles ecográficos…
Posteriormente, son necesarios cuidados neonatológicos esenciales: información y orientación a la familia; movilización precoz del paciente para evitar la tendencia a la ostopenia y a las fracturas repetitivas.
Rehabilitación: La rehabilitación debe comenzarse pronto en los niños con O.I., ya que mantener un buen nivel funcional hace disminuir fracturas y fortalece huesos y músculos. Los periodos de inmovilización tras las fracturas, deben ser lo más cortos posibles y procurando que exista carga y bidepestación incluso en el postoperatorio, cuando lleven escayolas o con férulas.
Tratamiento médico: Existen medicamentos recomendados para corregir la osteopenia y disminuir el número de fracturas. Consiguen una disminución del número de fracturas y, con esto, una mejoría significativa en la calidad de vida del paciente.
La hormona de Crecimiento ocasiona un aumento del metabolismo óseo y de estatura sin disminución del número de fracturas, pero si mejora de actividad muscular y aumento de velocidad de crecimiento. Es de utilidad combinada con otros medicamentos.
Tratamiento quirúrgico: Consiste en el enclavado de los huesos largos afectados, fundamentalmente en miembros inferiores. El objetivo es mantener la alineación conseguida con las osteotomías realizadas en un hueso largo y aumentar la resistencia mecánica del hueso al sumarle la del clavo, evitando la tendencia a la osteoporosis, a la deformidad progresiva y a las fracturas, permitiendo una marcha y utilización precoz del miembro operado. Los clavos pueden ser de dos tipos: sólidos o telescópicos.
DESENLACE
A causa de la variabilidad individual de la OI, es imposible hacer afirmaciones generales sobre las perspectivas de una persona afectada. Se recomienda que busque consejo personal.
En muchos casos, la fragilidad disminuye después de la pubertad por razones todavía desconocidas.
El pronóstico vital depende de la gravedad de las complicaciones respiratorias asociadas a las malformaciones espinales.
Comentarios
Publicar un comentario